The therapeutic potential of induced pluripotent stem cell (iPSC)-derived cell and gene therapy products is enormous, yet commercial advancement and broad patient access have not been realized. Significant progress has been made in clinical studies for indications including retinal disease, Parkinson’s, heart failure and spinal cord injury. Despite years of innovation, no iPSC-based therapy has reached the market. This might not be due to a lack of scientific ambition, but rather to early decisions that fail to anticipate the regulatory complexity ahead.
When developers wait too long to formalize their chemistry, manufacturing and controls (CMC) strategy, they often discover too late that the groundwork they laid is no longer fit for purpose. Re-derivation, regulatory rejection, or limited territorial access are possible consequences. As a result, the case for adopting an early, risk-informed CMC strategy is not just a regulatory preference — it is a necessity.
Cell line development in iPSC
Making an iPSC-derived medicine from cells procured from a donor or patient requires very early processing steps to develop a cell line suitable for further manufacturing steps. This cell line development (CLD) includes steps to isolate and process the cell source, reprogramming using viral or non-viral methods to dedifferentiate the cells to an iPSC phenotype, selection of an authentic clone, and expansion to a seed stock suitable for establishing a master cell bank (MCB).
The CLD steps are not easily amenable to a process fully compliant with Good Manufacturing Practice (GMP). Unit operations may include single-cell printing to high-throughput plates, analysis of clonal outgrowth, and picking and analysis of clones with ideal phenotypic characteristics. These are non-routine, difficult-to-document procedures often carried out in open cell culture systems and equipment designed for R&D CLD activities that are acceptable for standard biologic drugs (e.g. recombinant antibodies). However, unlike biologics, the resulting cell lines will be the active ingredient of iPSC-derived products, with few opportunities for impurity removal and no options for viral clearance or sterilization (Table 1). Consequently, iPSC cell line development demands greater reliance on quality and safety assurance than standard biologics.
A regulatory landscape without consensus
One of the most challenging aspects of iPSC development is the lack of global regulatory alignment on how GMP applies to early CLD. Is it possible to apply some of the principles of GMP but outside a full GMP environment?
In the EU/EEA and the UK, the GMP guidance for advanced therapy medicinal products (ATMPs), including iPSC-derived products, is set out in EudraLex Volume 4 Part IV. This anticipates that all processing steps are undertaken in licensed GMP establishments. It explains that in exceptional circumstances, it may be acceptable for GMP manufacture to start with cells that have been processed outside a GMP environment, but only if it is impossible to replace with a GMP alternative, the risks have been analyzed and controlled, and there is prior agreement from the competent authority responsible for clinical trial or marketing authorization.
In January 2025, the European Medicines Agency (EMA) published a guideline on quality requirements for ATMPs in clinical trials accepting that iPSC CLD may occur before a clear product concept was present, and that the stringency of oversight and documentation may be “reduced.” Nevertheless, it restates the Part IV exceptional-circumstances rules.
Other territories have adopted the GMP guidance published by the Pharmaceutical Inspection Co-operation Scheme (PIC/S) and do not publish their own standalone guidance (e.g. Singapore, Switzerland, Australia, Thailand, Canada, New Zealand). The 2021 publication of PIC/S Annex 2a providing GMP guidance for ATMPs includes a requirement that “principles of GMP may be applied” to stages prior to MCB, a softening of approach that is not present in the equivalent European Part IV text.
In the EU, there are competent authorities in each member state that may have an independent view on the application of GMP to CLD. Some authorities, such as the Paul Ehrlich Institute in Germany, expect all processing steps to be undertaken by licensed GMP manufacturers. Others, including the UK’s MHRA and the U.S. FDA, accept early CLD outside of fully licensed GMP settings, provided appropriate controls are in place. Such controls should ensure that suitable materials are used in CLD. The FDA in a September 2022 Town Hall meeting of its Office of Tissues and Advanced Therapies (OTAT) explained that, in theory, R&D iPSC CLD followed by GMP MCB production may be acceptable, but specifically warned developers against using research-grade materials for iPSC CLD if a higher grade is available.
So what should a developer do when regulatory requirements diff er between territories? This inconsistency places developers in a difficult position. Some regulatory authorities interpret GMP requirements strictly and expect evidence that early processing met the required standard. Developers that follow a more permissive path, such as relying solely on R&D laboratory-derived iPSC lines, may successfully apply for early clinical trials in one territory, but may later face issues with stricter regulators in other territories for later trials or marketing applications. This scenario would necessitate time-consuming and costly repetition of CLD under stricter conditions.
TABLE 1
How production processes impact the need for increased QA during CLD

When the cell line is the platform
Unlike many other therapy types, iPSC programs often rely on foundational cell lines that support multiple products or indications. These platforms may involve repeated gene editing or transfection cycles, requiring successive stages of CLD and banking. This creates a complex chain of activity where early errors become embedded in the foundation of the entire program and product portfolio. Furthermore, for such a program, one might question the wisdom in repeatedly alternating between full-GMP for banking and GMP-principles for CLD (Figure 1).
The view of the FDA’s OTAT is that this complexity makes the stakes even higher, with each CLD process accumulating risks, thus demanding increased scrutiny and quality assurance. Work done to establish a platform seed lot or MCB may ultimately support an entire pipeline, not just a single product. If those early decisions are not acceptable to regulators later, the consequences are wider than many developers realize.
Understanding GMP principles in context
Regulators may accept that insisting on full GMP for iPSC CLD is not just impractical but also may hinder development of novel medicines with unbearable cost and delays. This in turn may lead to reduced patient access to disruptive technologies that may deliver treatments for unmet needs. Therefore, a balance must be struck.
The phrase ‘principles of GMP’ may create confusion. It may appear to be a softer standard, but this is not a shortcut; in a scientific advice meeting with an EU regulator principles of GMP was defined as “full GMP without the license.” Whatever the regulatory expectations, quality risk matters to both the success of the iPSC product and to patient safety and benefit. Principles of GMP should be used to address these risks.
Apply standards for material assessment and traceability, ensuring that there is sufficient control of sterility, absence of mycoplasma, controlled endotoxin and TSE contamination, and adopt a xeno-free process if possible. When using viral vectors in derivation and genetic modification, ensure absence of replication competency, both by design of the vector and in subsequent MCB testing. Ensure that during CLD there is clear segregation of cell lines, to prevent mix-up and cross contamination or carry over. The environment in which CLD occurs must control contamination, particularly viable particles that could affect the cell line. It is vital that rigorous records are made and kept of all processing activities. Even without formal GMP licensing, these expectations must be met in a way that is defensible to regulators.
FIGURE 1
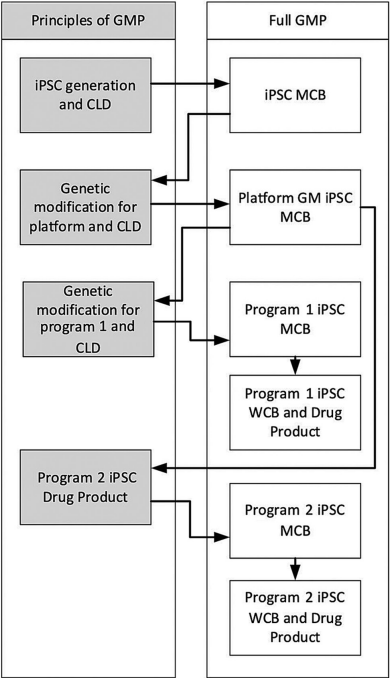
In a standard approach for iPSC-derived products that rely on a platform cell line that introduces an initial genetic modification and subsequent program-specific genetic modifications, the processing environment may alternate between the principles of GMP and full GMP, highlighting the need for sufficient control during CLD.
Developers should not assume an R&D process will be accepted; assess and document risks from the beginning. As an example, I advised a client against continuing cell line development in standard research laboratories due to insufficient control and monitoring and contamination risk. Instead, I recommended implementing documented cleaning and gowning procedures with environmental monitoring; although the space could not meet full EU GMP Grade A or B specifications at least the risks could be understood and assessed.
Early engagement matters
One of the most common mistakes iPSC-derived product developers make is delaying regulator engagement until nearing the point of clinical trial application. By that stage, the damage may already be done. Regulators may reject the cell source, the derivation process or the materials used, particularly if there is no pre-agreed justification for not meeting GMP principles.
Developers should consult with regulators before starting CLD, particularly if they plan to use GMP principles that fall well short of full GMP. If you want to take a less controlled approach, you need to get agreement from the authorities in advance. Do not seek a regulatory opinion after you have invested time and money into a process they may not accept.
What developers should do differently
There are several actions developers can take early in development to prevent delays and setbacks later:
- Conduct a formal risk-benefit assessment before initiating CLD
- Define a clear quality target product profile and a strategy that supports it, including early processing steps
- Document all materials, decisions and environmental controls, even in early research settings
- Consider the expectations of the most stringent regulators, not just the most convenient or lenient
- Engage regulators early and be transparent about planned deviations from full GMP
Regulators will listen to a well-reasoned strategy. But if you cannot provide the documentation or the scientific rationale, they will have no option but to reject your application.
Cell therapies derived from iPSCs face enough scientific and commercial challenges without adding avoidable regulatory barriers. Inconsistent global expectations, the risks of under-documented early work and the complexity of platform-based development all demand a shift in mindset.
A robust CMC strategy cannot be retrofitted; it should guide decisions from the very beginning. For developers aiming to bring iPSC-based therapies to the commercial market, the starting point is not the laboratory bench, it is the strategic plan.
Subscribe to our e-Newsletters
Stay up to date with news, articles and insights relevant to cell and gene therapy development and manufacturing. Plus, get special offers from Cell & Gene Therapy Review delivered right to your inbox!
Sign up now!