
As the most complex class of biologics therapeutics ever developed, cell and gene therapy (CGT) products face unique hurdles in transitioning from research lab processes and R&D to early clinical development manufacturing and then to commercial production and product launch. The key to maintaining a systematic approach in such a diverse class of therapeutics is to develop a control strategy for each of the innovative CGT therapies that will evolve throughout the product life cycle.
The present article describes the thought processes involved in defining control strategies across important areas including aseptic control, material control, process control, quality control and chain of custody control. We offer examples of how each of these elements should be developed and documented and discuss the connection of control strategy with product quality control and pharmaceutical quality systems. Additionally, we touch on future directions and the challenges of CGT manufacturing.
CGT as the new wave
Although not a brand-new therapeutic concept, autologous CAR-T product successes represent a new wave of complex biological therapeutics coming to maturity from an R&D concept to therapeutic reality. As CAR-T products achieved regulatory approvals, global excitement for CGT as a new class of biologics therapies was widespread, and within five years, the U.S., EU and China emerged as the biggest players in R&D and clinical development. The number of CGT programs in clinical trials rose year upon year since 2017, with slight dips during the COVID19 pandemic. By December 2023, there were 2,960 CGT clinical trials in progress in the world, with the promise of fulfilling unmet medical needs for human health care.
In spite of heavy investment in the CGT sector, manufacturing quickly proved to be vastly inadequate in terms of capacity, success rate and cost perspectives. Innovations in the design of the therapy to expand indications, improve gene modification delivery systems and increase quality control for this class of therapies pose problems for drug sponsors and regulators. CGT products all face commercial viability difficulties through supply or affordability issues stemming from high manufacturing and quality control costs that further limit wide adoption. CMC challenges have been identified as one of the biggest bottlenecks to the product and commercial success of this new class of promising therapeutics. A study that analyzed the 33 CGT clinical trials put on hold by the FDA from January 2020 to December 2022 showed that 21% of clinical holds were due to CMC issues.
Variability and CMC challenges
CGT therapies are highly complex therapies and the variability is great. Classifying sub-groups of CGT is no easy task as within and across each sub-type, derivatives may occur. For example, a cell therapy could also be a gene therapy when genetic modification of cells is involved; an iPSC therapy could be an autologous or allogeneic cell therapy differentiated into different cell types.
With such complexity, manufacturing process variability for this class of therapies is highly mutable. Conventional definitions for traditional biologics such as drug substance, drug product, or units of operations are not an easy fit for this new class of therapies. Likewise, critical raw material, manufacturing equipment and scales, and analytical methods must be carefully evaluated according to new criteria.
With the limited number and class of CGTs approved by the leading regulatory agencies, developers and manufacturers do not have enough public information to reference in designing their manufacturing processes and accompanying analytics and are even less likely to adopt a reliable control strategy for their CMC programs. The unknowns and higher risks of safety create uncertainty during product development. This is exacerbated by the fact that a large percentage of the CGT therapeutics obtain regenerative medicine advanced therapy (RMAT) designation or ‘Fast-Track’ status for clinical trials, putting CMC on the critical path for approval. In these scenarios, CMC mistakes or deficiencies are very costly in the time required to remedy the issue. The 2023 study previously cited found that CMC issues took an average of 8.4 months to resolve, much longer than the four to six months on average that it took to assess adverse events or address FDA requests for protocol amendments or preclinical information.
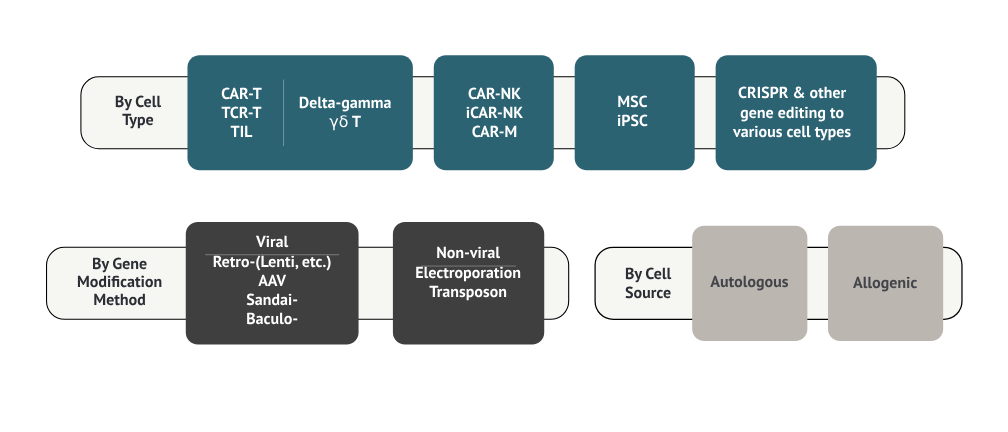
Figure 1. Classifying CGT products
Solutions for CGT manufacturing and quality
Closed systems for manufacturing
One obvious solution to the manual steps in CGT therapies typically achieved through single-use, presterilized components like bioreactors, tubing, filters, connectors, and strict environmental and personnel controls, is to create closed systems with automation capabilities. This means a completely contained, sterile production process where the therapeutic cells or viral vectors are handled within a sealed system throughout the entire manufacturing process, minimizing the risk of contamination by exposure to the outside environment, and operators, ensuring product quality and regulatory compliance. Automation with predefined parameters is the key to operate such closed systems.
Miltenyi Biotec, a leading cell therapy critical reagent provider, is among the first to embark on connecting various steps in a typical cell therapy production suite into one closed system and building automation to carry out the operational steps. Its automated cell processing platform, CliniMACS Prodigy Instrument, enables scalable GMP-compliant manufacturing of cell therapy products and is being used in a recent FDA approved CAR-T product from Autolus Therapeutics. Several other players dedicated to building solutions called ‘process in a box’ have emerged in recent years: Cellares Cell Shuttle claims to have integrated all unit of operations into a single cartridge, allowing 16 cartridges for multi-product, concurrent manufacturing, completed with integrated and automated industrial standard QC testing platforms. This has the potential to provide the most flexibility in a manufacturing facility with less footprint, fewer requirements for environmental controls and operator training, and offers significant cost savings compared to current commercially approved CAR-T therapy platforms. Another player to watch is OriBiotech. Their cell therapy manufacturing platform provides a rapid scale-out solution to manufacturers for whom carrying out similar processes for a large number of patients reliably and efficiently is a priority.
Product specific control strategy to unify quality management
Some common CMC challenges stem from small batch sizes and manual process steps required, making scale-up non-linear, even impossible in some cases. Using novel technology and equipment not previously designed for CGT may render process parameters difficult to control as the raw materials used tend not to be available in GMP grade and could introduce additional risks to the final product quality. Quality control for CGT is highly individualized and specific to the product and process: developers must justify QC testing from the sampling plan. Overall laboratory controls must be in order to render QC test results that meet data integrity requirements. CGT R&D and early clinical development largely come from academic and clinical research settings where adaptability to industrial/commercial settings are not considered. One example is the choice of cell lines and cell culture media:
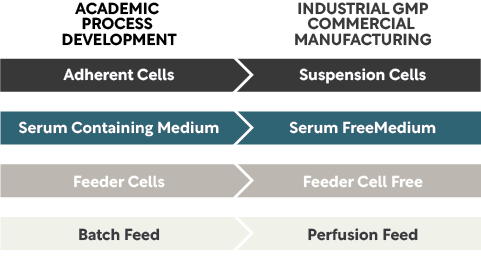
Figure 2: Cell therapy manufacturing cell line and culture media
Each CGT product is somewhat unique, and hence ‘platform knowledge’ for what constitutes critical quality attributes (CQA), or critical process parameters (CPP), that informs the strategy for controls is not available, including QC testing for each CGT product in clinical trial that the mature biologics can rely on. With very few regulatory approval precedents to follow, it is difficult to guess if the systems and controls you put in place for your CMC program are appropriate or adequate.
Control strategy
CMC programs tend to be on a critical path so careful, detailed consideration of manufacturing and product quality strategy must be in place before IND submission. FDA has published a guideline specific to CMC programs for CGT at the IND stage that highlights the critical areas. For most CGT developers, the big question is how do we know our process and quality controls are good enough for FDA/EMA to accept our IND application? The answer is — develop and start implementing your control strategy!
Control strategy, per ICHQ10 is defined as “A planned set of controls, derived from current product and process understanding that assures process performance and product quality. The controls can include parameters and attributes related to drug substance and drug product materials and components, facility and equipment operating conditions, in-process controls, finished product specifications, and the associated methods and frequency of monitoring and control.”
Control strategy is important for any drug to achieve the appropriate product quality, and essential for novel products/class of products to define and achieve CQA. The common CQAs for all biologics include bioburden, sterility, endotoxin, mycoplasma, and concentration of the active ingredients such as protein and cell number. The rest of the CQA are product-specific and must be defined and defended by the drug developers.
Control strategy is derived by first defining the quality target product profile (QTPP) of your CGT product, and a set of CQA corresponding to the QTPP. From there, each manufacturing process step, including the materials used for the process, and equipment capabilities needs to be evaluated from scientific and risk perspectives using data from process and analytical development, to arrive at the initial set of CPP and critical material attributes (CMA). This detailed analysis requires significant knowledge of the process and how to assess quality risks associated with each input.
Figure 3: Aspects of a control strategy
A standardized way to arrive at a control strategy
We maintain that a standard approach must be adopted to confidently design a control strategy and implement to phase 1 clinical production, then to evolve the strategy and implementation throughout clinical development phases until product launch. Do this by breaking down the control strategy into five sub-control strategies, as shown in Figure 3. This allows a way to assess potential impact to each of these aspects of the controls systematically while expecting certain overlaps in each type of control.
Aseptic controls usually involve:
- Clean rooms: Class B and Class A clean rooms are required for operations with any exposure to the environment; Class C clean rooms are required for closed system operations. They need to be validated and routinely cleaned and monitored, and maintenance of appropriate filters are also important.
- Aseptic operations: Training of manufacturing operators for aseptic techniques is an important GMP requirement.
- Material control and handling: Materials are a source of microbial input and therefore risk assessment including appropriate testing is required before they are released onto the manufacturing floor.
- In-process and final product testing: For microbials including bioburden, mycoplasma, endotoxin, adventitious viruses, and replication competent virus, testing must be performed when such contamination risks are present—which is the case for all biologics including CGT.
The material controls required for aseptic control is only one aspect to consider. Other controls may be required such as residuals of the materials used in manufacturing, for example, IL-2 and other stimulatory factors commonly added during the cell culture process. Each material used must be separately considered based on its function in the process, the concentration added, the known or unknown risks it may pose, how far is it from the final therapeutic product, and the ability of the process to reduce its amount in final product. The analysis for all the materials will be recorded in the control strategy for materials.
Quality control plays an even more critical role in CGT control strategy as the capabilities of the vast variety of processes are not yet proven. A robust QC system starting with a well-designed and maintained QC laboratory is the first step, with QC quality procedures in place to ensure proper control of the lab equipment, critical reagents, such as microbial strains and reference standards, and that analytical methods are suitable and qualified for the intended purposes and fully validated before final process validation. This demands a systematic approach and quality management expertise to lead the implementation. For CGT, significant challenges exist in tailoring the regulatory guidelines to a specific CGT product to demonstrate product safety with microbials and viruses and non-product residuals, and to design potency assays when the mechanism of actions are not well understood.
At the pre-IND stage, it is critical to record the knowledge and analysis that lead to the initial control strategy. This serves as justification for your manufacturing process descriptions, for QC testing schemes including sampling and justification of specifications for materials and product. The FDA has adopted a collaborative approach to regulation that relies on developers to provide knowledge and justification for the proposed control strategy. Quality by Design (QbD) is fully utilized to define acceptable operational spaces.
The critical link
Progress in biologics is rapidly being matched by progress in engineering. The variability and the novel nature of current CGT product development require a standard approach to defining controls in all aspects of manufacturing to ensure process and product quality. A well-developed control strategy based on phaseappropriate GMP principles must be developed before IND submission and implemented for initial clinical productions. The control strategy will evolve as the impact of various inputs to product quality, and in turn, clinical findings, are further understood. The changes must be documented in the developer’s quality system through change controls.
Developing a control strategy for a new CGT product requires bringing together substantial scientific, technical, GMP quality system knowledge and experience. A firm’s regulatory strategy and interactions with the regulators must be part of this systematic approach. To solve the CMC bottleneck for CGT product realization and commercial viability, control strategy is a critical link to exciting new manufacturing solutions for CGT that assure safety and efficacy of the product at different stages of the clinical development, leading to higher confidence in regulatory approval.
References
ClinicalTrials.gov A National Health Institute (NIH) sponsored searchable site for all clinical trials registered.
Wills C.A., Drago D., and Pietrusko RG. "Clinical holds for cell and gene therapy trials: Risks, impact, and lessons learned." Molecular Therapy: Methods & Clinical Development. Vol. 31. December 2023.
Chemistry, Manufacturing, and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs) Guidance for Industry
Pharmaceutical Quality System Q10, June 4th., 2008
Subscribe to our e-Newsletters
Stay up to date with news, articles and insights relevant to cell and gene therapy development and manufacturing. Plus, get special offers from Cell & Gene Therapy Review delivered right to your inbox!
Sign up now!